Studies of in vitro anti-prostate cancer potential of newer 1,2,4-triazolo-1,3,4-thiadiazines with different heteroaromatics
Abstract
This study was aimed to evaluate anticancer potential of newer synthesize 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazines and its derivatives. All newly furnished scaffolds were subjected to screening for their in vitro anticancer potential against DU-145 and PC-3 prostate cancer cell lines using SRB and MMT bioassays. The structures of final compounds were confirmed with the aid of FT-IR, 1H NMR, 13C NMR spectroscopy and CHN analysis. Bioassay studies suggested that all thiadiazines were promising cytotoxic agents with % cytotoxicity ranging from 44.39-71.24%, whereas potent GI50 level in the range 11.96-32.51 µg/mL and results were comparable to the potencies of control drugs adriamycin and doxorubicin. Variation of heterocyclic pharmacophores along with the C-5 position of 1,2,4-triazole in terms of quinoline, quinazoline, coumarin and pyridine lead to the different SAR predictions in which quinoline and benzimidazole moieties found most promising.
Introduction
Cancer therapeutics works upon inducing apoptosis and newer strategies to enhance apoptosis in cancerous cells upon targeting apoptosis regulators can be the vital factor in the further growth of anticancer regimen. Also, such newly invented scaffolds may combat drug resistant phenomenon towards the formally used chemotherapeutics (Earnshaw et al, 1999). In fact, nowadays, in clinics there is no selective cytotoxic agents available and current arsenal like hormone-therapy, radical prostatectomy and radiotherapy are successful in destroying androgen-dependent, localized prostate cancer growth, whereas, therapy of hormone-refractory prostate cancer remains restricted by unavoidable development of resistance to the first-line treatment with docetaxel drug. Consequently, novel medication is needed to create on immediate foundation to battle against extensive of this dangerous illness (Feldman and Feldman, 2001).
Heterocyclic compounds have long been known to exert desirable potency against several different diseases in terms of effective medications. Quinoline (Solomon and Lee, 2011), coumarin (Kostova, 2005) and benzimidazole (EL Rashedy, 2013) moieties as heterocyclic pharmacophore play a vital role in cancer drug discovery process. Moreover, the 1,2,4-triazole entity and their derivatives emerge rapidly with advance of modern heterocyclic chemistry, promising a variety of medical applications (Clemons et al, 2004; Wakale et al 2013). Furthermore, 1,3,4-thiadiazine ring system also contributed to the significant biological potential, in particular anticancer efficacies (Abdel-Rahman, 2006). Hence, in this study we have attempted the construction of new 1,3,4-triazolo-1,3,4-thiadiazines and screened their potential against prostate cancer cell lines.
Materials and Methods
TLC was performed with Merck precoated TLC plates, and the compounds were made visible using a fluorescent inspection lamp and iodine vapor. Gravity chromatography was done with Merck silica gel 60 (70-230 mesh). The melting points were recorded in open capillary tubes and are uncorrected. FT-IR spectra were recorded on FTIR Shimadzu (potassium bromide pellets) spectrometer (4000-400 cm-1). 1H and 13C NMR spectra were determined as DMSO solution at 400 and 100 MHz using TMS as an internal standard. Elemental analyses were performed on a Perkin Elmer EAL 240 elemental analyzer. All new compounds yielded spectral data consistent with the proposed structure and microanalysis within ± 0.4% of the theoretical values.
All hydrazide derivatives were synthesized according to the procedure reported in the literature for 2-(1H-benzo[d]imidazol-1-yl) acetohydrazide (1a) (Ansari et al., 2009; Patel et al., 2012), 2-oxo-2H-chromene-3-carbohydrazide (1b) (Bhat et al., 2008., Patel et al., 2013), quinoline-6-carbohydrazide (1c) (Saitoh et al 2009; Patel et al., 2012), 2-(6-bromoquinolin-4-yloxy) aceto hydra-zide (1d), 2-(1-methyl-2-oxo-1,2-dihydroquinolin-4-yloxy)acetohydrazide (1e), 2-(quinazolin-4-yloxy) aceto-hydrazide (1f) (Patel et al., 2012) and isoniazid or isonicotinohydrazide (1g) obtained commercially. Moreover, the formation of 3-(2-bromoacetyl)-2H-chromen-2-one via Knoevenagel condensation was also performed. All analytical data were in accordance with the literature data (Naik et al., 2013).
Synthetic procedure for synthesis of potassium aryldithio-carbazates (2a-2g)
0.2 Mol of derivatives 1a-1j was treated with a solution of 0.30 mol of KOH in 50 mL methanol at 0-5°C under N2 atmosphere with stirring. To this mixture, 0.30 mol of CS2 was added slowly and reaction mixture was allowed to stirred at RT overnight. The precipitated potassium aryldithiocarbazate was collected by filtration and washed with anhydrous ether and dried under vacuo. Potassium aryldithiocarbazate thus obtained was directly used in the next chemical procedure.
Synthetic procedure for synthesis of 4-amino-5-(substituted heteroaryl)-4H-1,2,4-triazole-3-thiols (3a-3g)
To a mixture of 0.1 mol of above potassium aryldithiocarbazates (2a-2g) in 10 mL of water, 0.3 mol of 99% hydrazine hydrate was added and reaction mixture was allowed to reflux for 8-11 hours until complete evolution of H2S gas and the color of mixture change to green and finally it became homogeneous. The reaction mixture was then cooled to RT and diluted with 100 mL of water and acidified with conc. HCl. The mixture was then extracted in EtOAC, washed with water, brine solution and dried over anhydrous Na2SO4. Pure 3a-3g were obtained using column chromatography with 5-6% EtOAc-petrolium ether as eluent as yellow solids.
Synthetic procedure for synthesis of title compounds 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazines (1-10)
A mixture of 4-amino-5-(substituted heteroaryl)-4H-1,2,4-triazole-3-thiols (3a-3g, 1 mmol) and 3-(2-bromoacetyl)-2H-chromen-2-one (1.1 mmol) in ethanol (5 mL) was refluxed for 6-13 hours. Upon completion of the reaction as monitored by TLC until no staring material left, the reaction mixture was concentrated and the residue thus obtained was diluted with water, extracted in EtOAc (75 mL). The combined extract was washed with 10% NaHCO3 solution, water followed by brine solution and dried over anhydrous Na2SO4. The crude product obtained was purified by column chromatography using 0.75-1.0% methanol in dichloromethane to afford the title compounds 1-10 (Puthiyapurayil et al., 2012).
The Scheme 1 outlines chemical reaction sequences and conditions required to furnish final compounds 1-10.
Scheme 1A: Synthetic protocol for 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazine analogues (1-10)
Scheme 1B: Synthetic protocol for 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazine analogues (1-10) (Cont.)
3-(3-((1H-benzo[d]imidazol-1-yl)methyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)-2H-chromen-2-one (1)
Yield 71%, m.p. 256-258°C, IR (KBr, cm-1): 3049 (Ar-H), 2988, 2919, 2845 (C-H), 1729 (lactone C=O), 1640 (C=N), 1389 (N=C-S), 1327 (C-N), 1317 (N-N=C), 679 (C-S-C bending); 1H NMR (400 MHz, DMSO-d6): d 8.49 (s, 1H, -N=CH, benzimidazole), 8.21 (s, 1H, H-4, coumarin), 7.83-7.67 (m, 2H, Ar-H), 7.35-7.43 (m, 2H, coumarin), 7.32-7.17 (m, 2H, Ar-H), 7.05-7.12 (m, 2H, coumarin), 4.21 (s, 2H, S-CH2, thiadiazine), 3.71 (s, 2H, -N-CH2); 13C NMR (100 MHz, DMSO-d6): δ 168.1 (1C, C=O, coumarin), 165.2, 162.6 and 160.1 (3C, C-3, C-6 and C-9, triazolothiadiazine), 153.2 (1C, C-9, coumarin), 152.7 (1C, C-2, benzimidazole), 145.2-116.8 (13C, Ar-C), 43.8 (1C, N-CH2), 41.2 (1C, C-7, CH2, triazolothiadiazine). Anal Calcd for C21H14N6O2S: C, 60.86; H, 3.40; N, 20.28; Found: C, 60.71; H, 3.58; N, 20.16.
3,3'-(7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine-3,6-diyl)bis(2H-chromen-2-one) (2)
Yield 68%, m.p. 245-257°C, IR (KBr, cm-1): 3058 (Ar-H), 2988, 2937, 2840 (C-H), 1734 (lactone C=O), 1633 (C=N), 1380 (N=C-S), 1331 (C-N), 1324 (N-N=C), 681 (C-S-C bending); 1H NMR (400 MHz, DMSO-d6): d 8.34 (s, 1H, H-4, coumarin), 8.37 (s, 1H, H-4, coumarin), 7.29-7.46 (m, 4H, coumarin), 7.04-7.15 (m, 4H, coumarin), 4.27 (s, 2H, S-CH2, thiadiazine); 13C NMR (100 MHz, DMSO-d6): d 169.1 (1C, C=O, coumarin), 167.8 (1C, C=O, coumarin), 166.1, 163.3 and 159.8 (3C, C-3, C-6 and C-9, triazolothiadiazine), 153.4 (1C, C-9, coumarin), 152.8 (1C, C-9, coumarin), 145.2-125.6 (14C, Ar-C), 41.9 (1C, C-7, CH2, triazolothiadiazine). Anal Calcd for C22H12N4O4S: C, 61.68; H, 2.82; N, 13.08; Found: C, 61.81; H, 2.97; N, 13.21.
3-(3-(quinolin-6-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)-2H-chromen-2-one (3)
Yield 64%, m.p. 288-290°C, IR (KBr, cm-1): 3042 (Ar-H), 2971, 2919, 2871 (C-H), 1724 (lactone C=O), 1648 (C=N), 1382 (N=C-S), 1324 (C-N), 1316 (N-N=C), 666 (C-S-C bending); 1H NMR (400 MHz, DMSO-d6): d 9.28 (dd, J = 2.1, 4.4 Hz, 1H, H-2, quinoline), 8.45-8.52 (m, 2H, H-3 and H-4, quinoline), 8.28 (s, 1H, H-4, coumarin), 8.09 (d, J = 1.9 Hz, 2H, H-5 and H-8, quinoline), 7.61 (dd, J = 4.8, 9.3 Hz, 1H, H-7, quinoline), 7.34-7.41 (m, 2H, coumarin), 7.08-7.17 (m, 2H, coumarin), 4.19 (s, 2H, S-CH2, thiadiazine); 13C NMR (100 MHz, DMSO-d6): d 169.2 (1C, C=O, coumarin), 165.7, 161.9 and 160.3 (3C, C-3, C-6 and C-9, triazolothiadiazine), 154.7 (1C, C-9, coumarin), 152.8, 151.1 (2C, C2 and C9, quinoline), 143.9-121.6 (14C, A-C), 43.7 (1C, C-7, CH2, triazolothiadiazine). Anal Calcd for C22H13N5O2S: C, 64.22; H, 3.18; N, 17.02; Found: C, 64.09; H, 3.37; N, 16.89.
3-(3-((6-bromoquinolin-4-yloxy)methyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)-2H-chromen-2-one (4)
Yield 59%, m.p. 277-280°C, IR (KBr, cm-1): 3045 (Ar-H), 2966, 2946, 2850 (C-H), 1730 (lactone C=O), 1644 (C=N), 1376 (N=C-S), 1325 (C-N), 1321 (N-N=C), 674 (C-S-C bending); 1H NMR (400 MHz, DMSO-d6): d 8.76 (d, J = 2.7 Hz, 1H, H-5, quinoline), 8.67 (d, J = 9.2 Hz, 1H, N=CH, H-2, quinoline), 8.31 (s, 1H, H-3, quinoline), 8.25 (s, 1H, H-4, coumarin), 8.21 (d, J = 7.5 Hz, 1H, H-8, quinoline), 8.15 (s, 1H, H-5, quinoline), 8.09 (dd, J = 8.3, 1.9 Hz, 1H, H-7, quinoline), 7.37-7.45 (m, 2H, coumarin), 7.05-7.11 (m, 2H, coumarin), 5.12 (2H, s, O-CH2), 4.26 (s, 1H, S-CH2, thiadiazine); 13C NMR (100 MHz, DMSO-d6): d 167.6 (1C, C=O, coumarin), 166.3, 163.1 and 161.6 (3C, C-3, C-6 and C-9, triazolothiadiazine), 152.8 (1C, C-9, coumarin), 152.2, 151.9 (2C, C2 and C9, quinoline), 151.3 (1C, O-CH2), 144.9-118.6 (14C, Ar-C), 39.9 (1C, C-7, CH2, triazolothiadiazine). Anal Calcd for C23H14BrN5O3S: C, 53.09; H, 2.71; N, 13.46; Found: C, 52.96; H, 2.89; N, 13.63.
1-methyl-4-((6-(2-oxo-2H-chromen-3-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-3-yl)methoxy)quinolin-2(1H)-one (5)
Yield 73%, m.p. 293-296°C, IR (KBr, cm-1): 3055 (Ar-H), 2969, 2933, 2864 (C-H), 1728 (lactone C=O), 1638 (C=N), 1380 (N=C-S), 1329 (C-N), 1318 (N-N=C), 684 (C-S-C bending); 1H NMR (400 MHz, DMSO-d6): d 8.21 (s, 1H, H-4, coumarin), 8.07 (d, J = 6.9 Hz, 1H, C-8 proton of quinoline), 7.72 (t, J = 8.2 Hz, 1H, C-7 proton of quinoline), 7.56 (d, J = 8.8 Hz, 1H, C-5 proton of quinoline), 7.47 (t, J = 6.6 Hz, 1H, C-6 proton of quinoline), 7.40 (s, 1H, C-3 proton of quinoline), 7.30-7.37 (m, 2H, coumarin), 7.04-7.09 (m, 2H, coumarin), 5.23 (2H, s, O-CH2), 4.22 (s, 2H, S-CH2, thiadiazine), 3.79 (s, 3H, N-CH3); 13C NMR (100 MHz, DMSO-d6): d 168.5 (1C, C=O, coumarin), 166.8 (1C, C=O, quinoline), 164.8, 161.9 and 159.5 (3C, C-3, C-6 and C-9, triazolothiadiazine), 155.0 (1C, C-9, coumarin), 151.9 (1C, O-CH2), 146.8-122.2 (16C, Ar-C), 42.6 (1C, C-7, CH2, triazolothiadiazine). Anal Calcd for C24H17N5O4S: C, 61.14; H, 3.63; N, 14.85; Found: C, 61.31; H, 3.51; N, 15.02.
3-(3-((quinazolin-4-yloxy)methyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)-2H-chromen-2-one (6)
Yield 66%, m.p. 259-261°C, 3039 (Ar-H), 2990, 2944, 2849 (C-H), 1722 (lactone C=O), 1640 (C=N), 1377 (N=C-S), 1334 (C-N), 1326 (N-N=C), 670 (C-S-C bending); 1H NMR (400 MHz, DMSO-d6): d 8.32 (s, 1H, H-4, coumarin), 8.22 (s, 1H, N-CH-N, quinazoline), 8.09 (dd, J = 10.2, 8.3 Hz, 2H, quinazoline ring), 7.33-7.41 (m, 2H, coumarin), 7.36-7.23 (m, 3H, Ar-H), 7.01-7.07 (m, 2H, coumarin), 5.17 (2H, s, O-CH2), 4.19 (s, 1H, S-CH2, thiadiazine); 13C NMR (100 MHz, DMSO-d6): d 167.1 (1C, C=O, coumarin), 165.7, 162.1 and 160.8 (3C, C-3, C-6 and C-9, triazolothiadiazine), 158.2, 157.7, 156.6 (3C, quinazoline), 153.8 (1C, C-9, coumarin), 144.6-124.7 (13C, Ar-C), 41.9 (1C, C-7, CH2, triazolothiadiazine). Anal Calcd for C22H14N6O3S: C, 59.72; H, 3.19; N, 18.99; Found: C, 59.87; H, 3.33; N, 19.13.
3-(3-(pyridin-4-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)-2H-chromen-2-one (7)
Yield 70%, m.p. 252-254°C, IR (KBr, cm-1): 3055 (Ar-H), 2977, 2917, 2848 (C-H), 1733 (lactone C=O), 1649 (C=N), 1391 (N=C-S), 1330 (C-N), 1320 (N-N=C), 681 (C-S-C bending);
1H NMR (400 MHz, DMSO-d6): d 8.87 (2H each, 2d, J = 5.8 Hz, 5.9 Hz, pyridine), 8.27 (s, 1H, H-4, coumarin), 7.38-7.44 (m, 2H, coumarin), 7.08-7.15 (m, 2H, coumarin), 4.21 (s, 2H, S-CH2, thiadiazine); 13C NMR (100 MHz, DMSO-d6): d 169.0 (1C, C=O, coumarin), 164.9, 161.8 and 161.0 (3C, C-3, C-6 and C-9, triazolothiadiazine), 154.5 (1C, C-9, coumarin), 150.3 (2C, pyridine), 143.7-123.9 (10C, Ar-C), 40.4 (1C, C-7, CH2, triazolothiadiazine). Anal Calcd for C18H11N5O2S: C, 59.82; H, 3.07; N, 19.38; Found: C, 59.69; H, 3.22; N, 19.25.
In vitro evaluation of anticancer activity
Sulforhodamine B (SRB) assay
Cancerous cell lines were grown using 2 mM L-glutamine and 10% fetal bovine serum under RPMI 1640 medium and incubation was carried out using 96-well microtiter plates having 90 µL at plating densities with regard to the doubling time of individual cell lines. At first, incubation of microtiter plates was performed at 95% air, 5% CO2, 100% relative humidity and 37 ± 1°C temperature for 24 hours before introducing the compounds and after inoculation. After incubation, for each line of cells, cell population was recorded on the time title compounds' appropriate concentrations (prepared using DMSO at 400-fold) were supplied (Tz) fixing one plate of each cell line with TCA. To the appropriate well 100 mL of each screen compound was inserted which already hold 100 mL of medium containing the cells which is to be incubated at 37°C for 48 hours (Skehn et al., 1990). Cells were fixed in situ by the soothing inclusion of 50 mL of cold 10% w/v TCA and incubated for 60 min at 4°C and then plated were cleaned properly and air dried. Then, 50 µL of 0.4% w/v SBR solution in 1% CH3COOH was added to each well and incubated for 20 min at 25°C and after incubation, plates were washed gently and air dried. Trizma base (10 mM) was used to dissolve the bound stain and the absorbance was read on an Elisa plate reader (690 nm reference wavelength) at a wavelength of 515 nm. %Growth was observed with reference to the control well for each plate and determined as the ratio of average absorbance of the test well to the average absorbance of the control wells X 100. Using the six absorbance measurements [time zero (Tz), control growth (C), and analyze development in the existence of drug at the four concentration levels (Ti), the percentage growth was calculated at each of the drug concentration levels. Percentage growth inhibition was calculated as: [(Ti-Tz)/(C-Tz)] x 100 for concentrations for which Ti > Tz and [(Ti-Tz)/Tz] x 100 for concentrations for which Ti<Tz. For the each test compound, the dose parameters were calculated in which GI50 level was calculated using the equation [(Ti-Tz)/(C-Tz)] x 100 = 50 which reveals the drug concentration resulting in a 50% reduction in the net protein increase (as measured by SRB staining) in control cells while the drug incubation. The TGI level which expressed the tested drug concentration resulting in total growth inhibition was determined from Ti=Tz. LC50 values expressed concentration of drug resulting in a 50% reduction in the measured protein at the end of the drug treatment as compared to that at the beginning and indicating a net loss of cells was calculated from [(Ti-Tz)/Tz] x 100=-50.
Cytotoxicity evaluation
MTT assay
MTT assay was conducted according to the process given in the literary works (Bopp and Lettieri, 2008) using control adriamycin. 96-Well plate was used to seed the cells followed by incubation for 24 hours at 37°C. The specific levels of examined substances (100 mg) were added in the wells of the plate. Controls were maintained to figure out the control cell survival (CS) and the percentage of live cells after culture. After incubation, the medium eliminated and 20 µL of MTT reagent was included in each dish followed by incubation for 4 hours at 37°C. The wells for formazan crystal formation development were noticed under microscopic lense. The yellowish MTT is reduced to dark colored formazan by viable cells only. After the removal of medium, 200 µL of DMSO was added and incubated at 37°C for 10 min. The color developed was quantified with an ELISA plate reader (570 nm). The CS is expressed as percentage (T/C x 100), where T = test, C = average of control readings. %Cytotoxicity was calculated by: %Cytotoxicity = 100 - %Viability.
Result and Discussion
Many wider factors of heterocyclic chemistry are identified as disciplines of general significance that impinge on almost all factors of modern organic and medicinal chemistry (Thiel, 2013). Moreover, 1,2,4-triazole nucleus has been previously emerged as promising lead to develop anti-cancer scaffolds (Kaczor et al., 2013; Zhao, 2012; Kumar et al., 2011). In this study, newer 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazines bearing different heteroaryls in their core structure were yielded via efficient chemical synthesis strategies. Correct synthesis of all intermediate compounds was confirmed from the literature report and all data were in accurate accordance with the proposed structures. In case of final compounds, the FT-IR data demonstrated the corresponding peak for C=N at 1633-148 cm-1, N=C-S peak at 1376-1391 cm-1, C-N peak in the range 1324-1334 cm-1, N-N=C band nearby 1316-1326 cm-1 as well as C-S-C bending was observed at 666-684 cm-1 which confirmed the correct formation of 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazine ring system. In addition, 1H NMR spectra of the final compounds were in accordance with the proposed structure as proton atoms of the coumarin nucleus resonated at around 8.21-8.37 ppm for H-4 atoms as well as other proton atoms as multiplets in the ranges around 7.29-7.46 and 7.01-7.17 ppm, while S-CH2 proton atoms from thiadiazine ring resonated around 4.19-4.27 ppm. Moreover, 13C NMR spectra gave accurate proof of correct formation of titled compounds as lactone carbon as C=O resonated at around 169 ppm in addition to the characteristic carbon atoms peaks in the region 160 ppm to 165 ppm for triazolothiadiazine as well as nearby 40 ppm. All of the novel compounds gave C, H and N analyses within 0.04% points from the theoretical values. The structures of the new compounds were fully supported through their spectroscopic data (FT-IR, 1H NMR, 13C NMR spectroscopy and CHN analysis).
Investigation of in vitro anti-cancer potential of titled compounds 1-10 was carried out against two prostate cancer cell lines PC-3 and DU-145 and bioassay results are summarized in Table I and results are expressed in terms of %cytotoxicity and GI50 values. From the results furnished, it can be stated that all final compounds are noticeable anti-cancer agents against targeted cell lines with MICs ranged from 11.96-32.51 µg/mL and 16.54-27.66 µg/mL against DU-145 and PC-3 cell lines, respectively. Overall, it was observed that varying pharmacophore at C-5 position of 1,2,4-triazole nucleus significantly affects the anti-cancer potential of resultant analogues. For instance, final analogues with quinoline ring system proved to be most potent against both cancerous cell lines in terms of %cytotoxicity (44.39-71.24%) followed by benzimidazole, quinazoline, coumarin and lastly pyridine ring system when compared to control drug Adriamycin with 80.55% of cytotoxicity level. It was inspected that final compounds were more active against DU-145 cell lines than PC-3 cell line except a compound bearing coumarin entity. In case of exerting anti-cancer potential in terms of GI50 level, almost similar trend was observed as %cytotoxicity, as 6-bromoquinoline ring containing final analogue was most potent followed by benzimidazole, other quinolines, quinazoline and pyridine rings system against DU-145 cell line. However, final analogue with benzimidazole nucleus was more potent than 6-bromoquinoline holding compound against PC-3 cell line. However, Pokhodylo et al , 2013 suggested that the combination of both 1,2,3-triazole and quinoline rings in one molecule resulted in interesting antitumor activity.
Table I: Anti-prostate cancer cell lines (PC3 and DU 145) activity of final compounds 1-7
| Entry | -R | MICs for PC3 cell line/DU-145 cell line | ||
|---|---|---|---|---|
| MTT assay | SRB assay | SRB assay | ||
| %Cytotoxicity | GI50 (µM) | GI50 (µg/mL) | ||
| 1 | 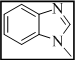 |
63.7 | 39.9/30.9 | 16.5/12.8 |
| 2 | 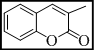 |
54.2 | 64.3/75.9 | 27.6/32.5 |
| 3 | 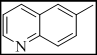 |
61.3 | 44.9/51.2 | 18.5/20.1 |
| 4 | 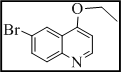 |
71.2 | 34.8/23.0 | 18.1/12.0 |
| 5 | 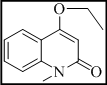 |
66.6 | 43.3/35.8 | 20.4/16.9 |
| 6 | 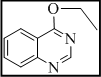 |
60.9 | 54.3/48.7 | 24.0/21.6 |
| 7 | 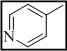 |
44.4 | 76.5/69.3 | 27.7/25.1 |
| Adriamycin | - | 80.6 | ||
| Doxorubicin | - | - | 0.4 | 0.4 |
From the bioassay it can be said that the overall potency of compound 4 with quinoline ring bearing electron withdrawing bromine atom was highest against both cell lines in terms of %cytotoxicty at 71.2% and GI50 level of 18.1 µg/mL and 12.0 µg/mL against PC-3 and DU-145 cell lines, respectively when compared to %cytotoxicty of control drug adriamycin at 80.6 and GI50 level of control drug doxorubicin at 0.4 µg/mL against both cell lines. Moreover, title compound 1 with benzimidazole nucleus showed 16.5 µg/mL and 12.8 µg/mL of GI50 level against PC-3 and DU-145 cell line, respectively, while its %cytotoxicty was recorded at 63.7%. These two derivatives were considered as most potent analogues among all tested. In addition, it can be said that insertion of quinoline ring system was the most beneficial part of the work carried out as compounds 3 and 5 exhibited 18.5 and 20.1 as well as 20.4 µg/mL and 16.9 µg/mL of GI50 levels against PC-3 and DU-145 cell lines, respectively. Overall all compounds were more active against DU-145 cell line with one exception as compound 2 with two coumarin rings systems furnished 27.6 and 32.5 µg/mL of GI50 level against PC-3 and DU-145 cell lines. From this particular result, it can be stated that further development of coumarin based 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazines may results into increased potencies of compounds against PC-3 cell line. In general, all compounds revealed noticeable action against PC-3 and DU-145 prostate cancer cell lines in both MTT and SRB bioassay as compared to the potencies of control drugs adriamycin and doxorubicin.
To sum up, quinoline ring with bromine atom was most vital part of the scaffold rationalization followed by benzimidazole, other unsubstituted quinolines, quinazoline and pyridine nucleus. All compounds except one with coumarin moiety demonstrated more inhibitory potential against DU-145 cell line than PC-3 cancerous cells and results are comparable with control drugs adrimycin and doxorubicin.
References
Abdel-Rahman TM. Synthesis, reactions, and anticancer activity of some 1,3,4-thiadiazole/thiadiazine derivatives of carbazole. Phosphorus Sulfur Silicon Related Elements. 2006; 181: 1737-54.
Ansari KF, Lal C. Synthesis and evaluation of some new benzimidazole derivatives as potential antimicrobial agents. Eur J Med Chem. 2009; 44: 2294-99.
Bhat MA, Siddiqui N, Khan SA. Synthesis of novel 3-(4- acetyl-5h/methyl-5-substituted phenyl-4,5-dihydro-1,3,4-oxadiazol-2-yl)-2h-chromen-2-ones as potential anticonvulsant agents. Acta Pol Pharm. 2008; 65: 235–39.
Bopp SK, Lettieri T. Comparison of four different colorimetric and fluorometric cytotoxicity assays in a zebrafish liver cell line. BMC Pharmacol. 2008; 8: 8.
Clemons M, Coleman RE, Verma S. Aromatase inhibitors in the adjuvant setting: Bringing the gold to a standard. Cancer Treat Rev. 2004; 30: 325-32.
Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: Structure, activation, substrates, and functions during apoptosis. Ann Rev Biochem. 1999; 68: 383-424.
El Rashedy AA, Aboul-Enein HY. Benzimidazole derivatives as potential anticancer agents. Mini Rev Med Chem. 2013; 13: 399-407.
Feldman BT, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001; 1: 34-35.
Kaczor AA, Pitucha M, Karczmarzyk Z, Wysocki W, Rzymowska J, Matosiuk D. Structural and molecular docking studies of 4-benzyl-3-[(1-methylpyrrol- 2-yl)methyl]-4,5-dihydro-1H-1,2,4-triazol-5-one with anticancer activity. Med Chem. 2013; 9: 313-28.
Kostova I. Synthetic and natural coumarins as cytotoxic agents. Curr Med Chem Anticancer Agents. 2005; 5: 29-46.
Kumar D, Narayanam MK, Chang KH, Shah K. Synthesis of novel indolyl-1,2,4-triazoles as potent and selective anticancer agents. Chem Biol Drug Des. 2011; 77: 182-88.
Naik RJ, Kukkarni MV, Palakshammurthy BS, Devarajegowda HC. Non-sulfur containing Z-1-chloro-3-coumarinyl 2-bromoethylenes from chlorosulfonation of 3-bromoacetyl coumarins. Indian J Chem B. 2013; 52: 1468-72.
Patel RV, Kumari P, Chikhalia KH. New quinolinyl–1,3,4–oxadiazoles: Synthesis, in vitro antibacterial, antifungal and antituberculosis studies. Med Chem. 2013; 9: 596-607.
Patel RV, Kumari P, Rajani DP, Chikhalia KH. Synthesis of coumarin-based 1,3,4-oxadiazol-2ylthio-N-phenyl/benzothiazolyl acetamides as antimicrobial and antituberculosis agents. Med Chem Res. 2013; 22: 195–210.
Patel RV, Patel PK, Kumari P, Rajani DP, Chikhalia KH. Synthesis of benzimidazolyl-1,3,4-oxadiazol-2ylthio-N-phenyl (benzothiazolyl) acetamides as antibacterial, antifungal and antituberculosis agents. Eur J Med Chem. 2012; 53: 41-51.
Patel RP, Patel JK, Kumari P, Chikhalia KH. Combination of bioactive moieties with different heteroatom(s): Application of Suzuki cross coupling reaction. Heteroatom Chem. 2012; 23: 399-410.
Pokhodylo N, Shyyka O, Matiychuk V. Synthesis of 1,2,3-triazole derivatives and evaluation of their anticancer activity. Sci Pharm. 2013; 81: 663-76.
Puthiyapurayil P, Poojary B, Chikkanna C, Buridipad SK. Synthesis, spectral characterization and biological evaluation of a novel series of 6-arylsubstituted-3-[2-(4-substitutedphenyl)propan-2-yl]-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines. Eur J Med Chem. 2012; 57: 407-16.
Saitoh M, Kunitomo J, Kimura E, Hayase Y, Kobayashi H, Uchiyama N, Kawamoto T, Tanaka T, Mol CD, Dougan DR, Textor GS, Snell GP, Itoh F. Design, synthesis and structure-activity relationships of 1,3,4-oxadiazole derivatives as novel inhibitors of glycogen synthase kinase-3β. Bioorg Med Chem. 2009; 17: 2017-29.
Skehn R, Storeng A, Scudiero J, Monks D, McMohan D, Vistica TW, Jonathan H, Bokesch S, Kenney MR, Boyd J. New colorimetric cytotoxicity assay for anticancer-drug screening. Natl. Cancer Inst. 1990; 82: 1107.
Solomon VR1, Lee H. Quinoline as a privileged scaffold in cancer drug discovery. Curr Med Chem. 2011; 18: 1488-508.
Thiel O. Heterocyclic chemistry in drug discovery. Li JJ (ed). Wiley, 2013, p 52.
Wakale VS, Pattan SR, Vishal T. Therapeutic importance of 1, 2, 4-triazole: A review. Int J Res Pharmaceut Biomed Sci. 2013; 4: 985-1001.
Zhao PL1, Duan AN, Zou M, Yang HK, You WW, Wu SG. Synthesis and cytotoxicity of 3,4-disubstituted-5-(3,4,5-trimethoxyphenyl)-4H-1,2,4-triazoles and novel 5,6-dihydro-[1,2,4]triazolo[3,4-b][1,3,4] thiadiazole derivatives bearing 3,4,5-trimethoxyphenyl moiety. Bioorg Med Chem Lett. 2012; 22: 4471-74.

