Synthesis of isatin, 5-chloroisatin and their ∆-2-1, 3, 4 oxadiazoline derivatives for comparative cytotoxicity study on brine shrimp
Abstract
Isatin (3a), isatin 3-carbohydrazone (4a), 5-spiro (isatin) 2-(N-acetyl hydra-zino) 4-(N-acetyl)- ∆2-1,3,4 oxadiazoline (5a), and 5-spiro (isatin) 2-hydrazino- ∆2-1,3,4 oxadiazoline (6a) had been synthesized from the unsubstituted oximinoacetanilide (2a). 4-Chlorooximinoacetanilide (2b), 5-chloroisatin (3b), 5-chloroisatin 3-carbohydrazone (4b) and 5-spiro (5-chloroisatin) 2-(N-acetyl hydrazino) 4-N-acetyl ∆-2-1,3,4 oxadiazoline (5b) compounds had been synthesized from p-chloroaniline. The structures of the products had been characterized from the spectral analysis and comparative cytotoxicity study of them was studied.
Introduction
Isatin, possessing an indole nucleus having both the keto and lactam moiety has aroused tremendous curiosity due to its diverse biological and pharmacological studies. From literature survey it is well known that isatin heterocycles exhibit manifold importance in the field of medicinal chemistry as a potent chemotherapeutic agent. Recently Islam et. al., (1992, 2001), in collaboration with National Cancer Institute (NCI) of USA, it was observed that acylated ∆2-1,3,4 thiadiazoline derivatives of isatin showed effective anticancer activity against a number of cancer cells especially for breast cancer. This eventful observation encouraged us to devotion of further research on isatin heterocycles reacting with carbohydrazide, especially for spiro 1, 3, 4 oxadiazoline derivatives. We report herein the synthesis of compounds shown in the scheme 1 following previous literature (Islam et al., 2001). The synthesized compounds were characterized by spectral analysis of IR, 1H-NMR and mass spectrometry. The result of the systematic study on cytotoxicity of the synthesized compounds on brine shrimp has also been discussed.
Scheme 1
Materials and Methods
The melting points of the synthesized compounds were recorded by thin disc method on a FISCHER JOHN'S electrothermal melting point apparatus. TLC was used for the monitoring the progress of reactions. Infrared spectra were recorded on DR-8001, SHIMADZU FT-IR spectrophotometer as a solid which was finely grounded in a small agate mortar in KBr disc. 1H-NMR spectra were measured by WP 200-NMR spectrometer using TMS (tetramethyl silane) as an internal standard and DMSO-d6 (dimethyle sulphoxide) as a solvent. Mass spectra were recorded on a high resolution mass spectrometer, KARATAS MS-25 using DH-88 data system.
Synthesis of isatin, 3a: Following the previous literature (Islam et al., 2001) oximinoacetanilide was prepared in 75% yield. After addition of concentrated sulphuric acid (14.7 mL) with dry oximinoacetanilide (4 g; 0.024 mole) at 70-80°C the solution was cooled to room temperature and poured upon 10-12 times of its volume of crushed ice. The orange red crude solid product of 3a was separated from the solution after half an hour which was filtered, washed well with cold water and dried in a vacuum desiccators. The orange red pure product of 3a was recrystallized from ethyl acetate having m.p. 180-182°C, yield 2.3 g (65%); Rf 0.4 (PE : EA ; 3 : 2).
IR: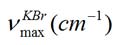 : 3189 (br,nNH; amide), 3106 (w,nCH, aromatic), 1726 (sh,nC=O, keto), 1614 (sh,nC=O, lactam), 1460 (sh,nC-C, aromatic).
: 3189 (br,nNH; amide), 3106 (w,nCH, aromatic), 1726 (sh,nC=O, keto), 1614 (sh,nC=O, lactam), 1460 (sh,nC-C, aromatic).
1H-NMR (DMSO-d6): d ppm: 11.02 (s, 1H; NH); 6.90 (d, J=7.8 Hz, 1H, C1-H); 7.12 (t, 1H, C2-H); 7.56 (t, 1H; C3-H); 7.49 (d, J=7.8 Hz, 1H, C4-H).
MS m/z (% of relative intensities): 147 (M+, 70), 119 (100), 92 (68), 76 (11), 74 (6) and 64 (32). M+ represents the molecular formula of C8H5NO2.
Synthesis of isatin 3-carbohydrazone, 4a: The reaction mixture of carbohydrazide (1 g, 6.8 mmol) in glacial acetic acid (31.6 mL) and a hot solution of isatin (0.6 gm; 6.8 mmol) in glacial acetic acid (33.1 mL) was refluxed for 3-4 hours with vigorous stirring which afforded a crude mass of 4a. When the reflux was completed, the reaction mixture was cooled, filtered and the crude product was washed well with water. Recrystallization of the crude 4a from methanol gave a yellow colored pure compound of 4a having m.p. >3000C, yield 1.0 g (70%) and Rf 0.3 (PE: EA; 2:1)
IR:: 3395 (w,nNH; 10 amine; symmetric), 3305 (w,nNH, 10 amine; asymmetric), 3198 (br,nNH, 20 amine), 3058 (w,nCH, aromatic), 1730 (sh, nC=O, keto), 1685 (sh,nC=O, lactam), 1624 (sh, nC=N), 1513 (sh, nC-C, aromatic).
1H-NMR (DMSO-d6): d ppm: 11.29 (s, 3H; 3NH); 7.58 (d, J=7.8 Hz, 1H; C1-H); 7.42 (m, 1H, C3-H); 7.12 (t, 1H; C2-H); 7 (d, J=7.8 Hz, 1H, C4-H).
MS m/z (% of relative intensities): 219 (M+, 10), 207 (13), 161 (72), 144 (18), 133 (16), 116 (12), 104 (42), 90 (19) and 76 (26). M+ represents the molecular formula of C9H9N5O2.
Synthesis of 5-spiro (isatin) 2(N-acetyl hydra-zino)-4-(N-acetyl)-∆2-1,3,4-oxadiazoline, 5a: The oxidative cyclization of isatin 3-carbohydrazone 4a (0.4 gm; 2 mmol) with freshly distilled acetic anhydride (55 mL) by refluxing (Kubota et al., 1980) for four hours led to the synthesis of the compound, 5a. The crude solid mass of 5a was purified by recrystallization from ethyl acetate and orange crystalline solid of 5a was obtained having m.p. 120-122°C, yield 460 mg (80%) and Rf 0.6 (EA : PE ; 1:4).
IR:: 3259 (sh,nNH), 3132 (sh,nCH, aromatic), 2932 (sh,nCH, aliphatic), 1785 (sh, nCOCH ), 1705 (sh,nC=O,keto), 1660 (sh,nC=N), 1609 (sh,nC-C, aromatic).
1H-NMR (DMSO-d6): d ppm: 12.15 (br.s, 3H; 3´NH); 8.17 (d, J=7.8 Hz, 1H, C1-H); 7.73 (d, 1H; d); 7.51-7.67 (m, 1H; C3-H); 7.41 (t, 1H; C2-H); 2.6 (s, 3H, -NCOCH3); 2.17 (s, 3H; - NHCOCH3).
MS m/z (% of relative intensities): 288 (M+-15, 5), 245 (7), 202 (10), 175 (70), 160 (5), 146 (6), 132 (11), 104 (11), 90 (8) and 77 (9)
Synthesis of 5-spiroisatin 2-hydrazino ∆2-1,3,4 oxadiazoline 6a: The reaction of 5-spiro (isatin) 4-(N-acetyl)-2(N-acetyl amino) D2-1,3,4-oxadiazoline, 5a (0.2 g, 1 mmol) and hydrazine hydrate (2.5 mL, 98%) with stirring at room temperature for 6 hours afforded precipitate of the crude product of 6a. The resulting precipitate was filtered, dried in a vacuum desiccators and recrystallized from water and obtained pale yellow powdered solid of 6a having m.p. 210-212°C, yield 69.7 mg (40%) and Rf 0.2 (EA : PE ; 1: 4)
IR:: 3358 (sh,nNH), 3158 (br,nCH, aromatic), 1686 (sh,nC=O, lactam), 1657 (sh,nC=N), 1588 (sh,nC-C, aromatic).
1H-NMR (DMSO-d6) : d ppm : 11.84 (br.s, 1H, N1-H, lactam); 11.28 (s, 1H, N2-H); 10.84 (br.s, 1H, N3-H); 7.56 (d, J=7.8 Hz., 1H, C1-H), 7.37 (t, 1H, C2-H), 7.09-7.16 (m, 1H; C3-H); 7.00 (d, J=7.8 Hz., 1H, C4-H).
MS m/z (% of relative intensities): 204 (M+-15, 10), 149 (14), 138 (8), 119 (6.6), 97 (5.83), 83 (9.58), 55 (40), 43 (96), 28 (100) and 17 (31).
Synthesis of p-chlorooximinoacetanilide, 2b: In accordance with the modified Sandmeyer procedure (Islam et. al., 1997) compound 2b was prepared from p-chloroaniline (8 gm, 0.1 mole), chloral hydrate (10.9 gm, 0.1 mole), saturated aqueous solution of sodium sulfate (157.8 mL), concentrated hydrochloric acid (5.2 mL) and hydroxylamine hydrochloride (13.4 gm, 0.2 mole). The crude product obtained as a pale brown solid which on recrystallization from ethyl acetate yielded brown colored powdered solid of 2a having m.p. 170-172°C, Rf 0.5 (PE:EA, 2:1) and yield 1.0 g (80%).
IR:: 3303 (sh,nOH), 3203 (sh,nNH), 3106 (sh,nCH, aromatic), 1663 (sh,nCO, amide), 1623 (sh,nC=N), 1569 (sh,nC-C, aromatic).
1H-NMR (DMSO-d6): d ppm: 12.2 (s, 1H, N-OH), 10.2 (s,1H, NH), 7.62 (s, 1H, CH=N-), 7.71 (d, J=7.6 Hz., 1H, H-3, Ar-H), 7.36 (d,J=7.6 Hz., 1H, H-4, Ar-H).
MS m/z (% of relative intensities): 198/200 (M+ /M++2, 3:1, 35Cl, 80), 127/129 (35Cl, 100), 181/183 (35Cl, 10), 167/169 (35Cl, 4), 153/155 (35Cl, 26), 144/146 (35Cl,12), 111/113 (35Cl, 11), 99/101 (35Cl, 24), 90/92 (35Cl, 10) and 75/77 (35Cl, 16).
Synthesis of 5-chloroisatin, 3b: The cyclisation of p-chlorooximinoacetanilide (6 gm, 0.03 mole) according to standard procedure of Sandmeyer treated with concentrated H2SO4 (22.0 mL) on controlled temperature led to the synthesis of 5-chloroisatin. The crude product of 3b was obtained as a pink-red solid which on recrystallization from ethyl acetate afforded brick-red powdered solid having m.p. 230-2320C, Rf 0.4 (PE:EA, 1:1) and yield 3.8 g (70%).
IR:: 3097 (w,nNH), 3058 (w,nCH, aromatic), 1704 (sh,nC=O, keto), 1617 (sh,nC=O, lactam), 1470 (sh,nC-C, aromatic).
1H-NMR (DMSO-d6): d ppm: 11.12 (s, 1H, NH, lactam), 6.91-6.95 (d, J=7.6 Hz., 1H, H-3, Ar-H), 7.55-7.61 (t, 1H, H-2, Ar-H).
MS m/z (% of relative intensities): 181/183 (M+ /M++2, 3:1, 35Cl, 48), 153/155 (35Cl, 100), 125/127 (35Cl, 34), 110/112 (35Cl, 7), 98/100 (35Cl, 15), 90 (35Cl, 16), 75 (35Cl, 10), 63 (35Cl, 38).
Synthesis of 5-chloroisatin 3-carbohydrazone, 4b: The refluxing of unimolecular ratio of 5-chloroisatin, 3b (1.2 gm, 6.6 mmol in 38.0 mL glacial acetic acid) and carbohydrazide (0.6 g, 6.6 mmol in 32.2 mL glacial acetic acid) for four hours afforded precipitate of 4b. After usual workup bright yellow solid of 4b was obtained by recrystallization from methanol; m.p. >300°C, Rf 0.3 (PE: EA, 1:1) and yield 1.1 gm (70%).
IR:: 3562 (sh,nNH , symmetric), 3415 (sh,nNH, asymmetric), 3345 (w,nNH), 3202 (br, nCH, aromatic), 1786 (sh,nC=O, keto), 1734 (sh,nC=O, lactam), 1620 (sh, nC=N), 1470 (sh,nC-C, aromatic).
1H-NMR (DMSO-d6): d ppm: 11.45 (s, 1H, NH, lactam), 8.55 (s, 2H, 2´NH), 7.4 (d, J=7.6 Hz., 1H, H-2, Ar-H), 7 (d, J=7.6 Hz., 1H, H-3, Ar-H).
MS m/z (% of relative intensities): No M+/ M++2 peak was observed due to loss of thirty mass unit. 223/225 (M+ -30/(M++2) -30, 3:1, 35Cl, 12), 207/209 (35Cl, 88), 195/197 (35Cl, 100), 178/180 (35Cl, 10), 151/153 (35Cl, 14), 138/140 (35Cl, 43), 124/126 (35Cl, 15), 112/114 (35Cl, 9), 102 (35Cl, 14), 88 (35Cl,11), 75 (35Cl, 21).
Synthesis of 5-spiro (5¢-chloroisatin)-2-(N-acetyl hydrazino)-4-(N-acetyl)-∆2-1,3,4 oxadiazoline, 5b: The refluxing of 5-chloroisatin 3-carbohy-drazone (0.3 g, 1.1 mmol) with freshly distilled acetic anhydride (84.2 mL) with vigorously stirring for two hours and usual workup led to a grayish solid of 5b. The product on recrystallization from dichloromethane afforded dark grayish solid of 5b having m.p. 174-175°C, Rf 0.4 (EA: PE, 3:2) and yield 253.7 mg (75%).
IR:: 3255 (sh,nNH), 2933 (w,nCH, aliphatic), 1780 (sh,nC=O, -NHCOCH3), 1724 (sh, nC=O, lactam), 1616 (sh,nC=N), 1591 (sh,nC-C, aromatic).
1H-NMR (DMSO-d6): d ppm: 12.11 (s, 3H, 3´NH), 8.15 (d, J=7.6 Hz., 1H, H-6, Ar-H), 7.82 (s, J=7.6 Hz., 1H, H-4, Ar-H), 7.65 (d, 7.5 Hz, 1H, H-3, Ar-H), 2.61 (s, 3H, terminal -NCOCH3), 2.19 (s, 3H, -NHCOCH3).
MS m/z (% of relative intensities): No M+/ M++2 peak was observed due to loss of fifteen mass unit. 322/324 (M+ -15/(M++2) -15, 3:1, 35Cl, 3.2), 321/323 (35Cl, 16), 279/281 (35Cl, 14), 251/253 (35Cl, 13), 236/238 (35Cl, 50), 194 (35Cl, 21), 180 (35Cl, 17), 166 (35Cl, 20), 138 (35Cl, 13), 102 (35Cl, 10).
Demonstration of cytotoxicity: The cytotoxicity study of the synthesized compounds - 3a, 4a, 5a, 6a, 2b, 3b, 4b and 5b was investigated on brine shrimp as a test organism for convenience. 1.6 mg of each of the compounds was taken in the corresponding sample vials with 1.6 mL of dimethyl sulfoxide to prepare stock solution. From this stock solution 33, 99 and 132 ppm of each compounds were placed in separate test tubes by micro syringe 1 mL of extra dimethyl sulfoxide was given in each test tube with 10-12 brine shrimp (Microwell, 1997). After 1, 2, 3 and 4 hours the test tubes were observed and the number of survived naupli in each test tube was counted and results were noted. From this the percentage of lethality of brine shrimp naupli was calculated at each concentration for each sample. Then graphs were drawn by plotting percentage of lethality of brine shrimp versus doses (in ppm) of the synthesized compounds which gave rise to the LD50 values of the corresponding compounds.
The LD50 values of the synthesized compounds are given in Table I.
Table I: LD50 values of the synthesized compounds
| Compound | LD50 | Remarks |
|---|---|---|
| 5b | 21 | Highly active |
| 5a | 24 | |
| 4a | 25 | |
| 3a | 27 | Medium active |
| 6a | 28 | |
| 2b | 32 | Weakly active |
| 4b | 33 | |
| 3b | 99 | Inactive |
Sample Number
The following graph represents the comparative LD50 values of the synthesized compounds (3a, 4a, 5a, 6a, 2b, 3b, 4b and 5b)
Result and Discussion
The structure of isatin (3a) has been confirmed by its IR, 1H-NMR and mass spectra. The IR bands at 3189 cm-1, 3106 cm-1, 1614 cm-1 and 1460 cm-1 correspond to the presence of broad band of NH, CH (aromatic), C=O (keto), C=O (lactam) and C-C (aromatic) groups respectively. In 1H-NMR a sharp singlet appearing at d 11.02 is due to NH proton (lactam). Four aromatic protons of C1-H, C2-H, C3-H, and C4-H appear at d 6.90 (d, J=7.8 Hz., 1H, C4-H), d 7.12 (t, N, C2-H), d 7.56 (t, C3-H) and d 7.49 (d, J=7.8 Hz., 1H, C4-H) respectively. Mass spectrum confirms the structure of 3a which shows molecular ion peak M+ at m/z 147.
Reaction of 3a with carbohydrazide in glacial acetic acid gave 4a, whose mass spectrum showed molecular ion peak at m/z 219 which is consistent with the structure of the compound. Acetylation of 4a with acetic anhydride afforded 5a. IR spectrum of 4a indicates the presences of acetyl group (-COCH3) by the band appearing at 1785 cm-1. 1H-NMR spectrum confirms the presence of two acetyl groups by two singlets at d 2.2 for NH-COCH3 and at d 2.6 due to -NCOCH3. The expected molecular ion peak (M+) was not found due to less of fifteen mass unit. Two NH protons were not observed in the 1H-NMR spectrum due to rapid proton-deuteron exchange reaction in deuterated dimethyl sulfoxide solvent.
Reduction of 5a by hydrazine hydrate stirring at room temperature afforded 6a. The IR spectrum indicated the absence of any acetyl group which confirmed the reduction of 5a has taken place. The bands at 3358 cm-1 and 1686 cm-1 indicate the presence of NH and C=O (lactam) groups respectively. In 1H-NMR a broad singlet appears at d11.84 due to NH proton of lactam, whereas a sharp singlet at d 11.28 and a broad singlet at d 10.84 indicate the presence of NH proton of N2-H and NH proton of N3-H respectively. No signal of two NH2 protons was observed due to rapid proton-deuteron exchange reaction in deuterated dimethyl sulfoxide solvent. In mass spectrum the molecular ion peak M+ was not observed due to loss of fifteen mass unit from 6a instead the peak appearing at m/z 204 (M+-15) is consistent with the structure of 6a.
The IR bands of 2b at 3303 cm-1, 3203 cm-1, 1663 cm-1 and 1623 cm-1 indicates the presence of -OH, -NH, C=O (amide) and C=N groups respectively. The 1H-NMR shows a doublet at d 7.7 due to the effect of neighboring electronegative chlorine of aromatic proton H-3 and a doublet at d 7.4 due to aromatic proton H-4. Two singlets appear at d 12.2 and at d 10.2 due to OH and NH proton respectively. In mass spectrum the molecular ion peak at m/z 198/200 (3:1) which is consistent with the isotopic pattern of chlorine and structure of 2b.
The IR spectrum of 3b showed bands at 3097 cm-1, 1704 cm-1 and 1617 cm-1 correspond to NH, C=O (keto) and C=O (lactam) groups respectively. The 1H-NMR shows a broad peak at d 11.1 due to NH proton. Aromatic proton H-2 undergoes coupling with H-3 and meta coupling with H-1 simultaneously and appears as a triplet at d 7.55-7.61. A doublet appears at d 6.91-6.95 due to aromatic proton of H-3. The molecular ion peak at m/z 181/183 (3:1) is characteristic of isotopic pattern of chlorine and consistent with the structure of 3b.
In IR spectrum of compound 4b, the bands appear at 3562 cm-1, 1786 cm-1 and 1620 cm-1 indicate the presence of -NH, C=O (keto) and C=N groups respectively. The 1H-NMR shows a sharp singlet at d 11.5 due to NH proton of lactam and a singlet at d 8.5 due to two NH protons but two NH2 protons were not observed due to rapid proton deuteron exchange reaction in deuterated dimethyl sulfoxide solvent. The mass spectrum did not show the expected molecular ion peak M+ due to loss of thirty mass unit and showed the peak at m/z 223/225 (M+-30, 3:1) which is characteristic of isotopic pattern of chlorine and consistent with the structure of 4b.
Acetylation of 4b with freshly distilled acetic anhydride gave 5b. The IR spectrum of 4b shows bands at 1780 cm-1 indicating presence of acetyl group. The 1H-NMR spectrum of 5b confirms the presence of two acetyl group groups by two sharp singlets at d 2.2 and at d 2.6 due to three protons of -NHCOCH3 and -NCOCH3 respectively. A broad singlet appears at d 12.1 represents three NH protons of H-4, H-5 and H-6. In mass spectrum the molecular ion peak M+ was not observed due to loss of fifteen mass unit instead of that a peak appeared at m/z 322/324 (3:1), which is characteristic of isotopic pattern of chlorine and consistent with the structure of 5b.
On the basis of cytotoxicity study of the synthesized compounds it was found that carbohydrazido group containing unsubstituted isatin activates the cytotoxic effect of the compound, than that of isatin itself. Spiro 1, 3,4 oxadiazoline derivatives of isatin (5b) having chlorine atom showed cytotoxic effect on brine shrimp more significantly than 5a without chlorine atom.
References
Islam MR, Khayer K, Shaha G, Chowdhury MSK. Syntheses of thiocarbohydrazide, some thiocarbohydrazones and their cyclized products as probes for pharmacological studies. Indian. J Chem. 1992; 31B: 547.
Islam MR, Khayer K, Abedin MJ. Reaction of isatin with 2-aminophenol leading to spiroheterocycles having anticancer activity. Jahangirnagar Univ J Sci. 2001; 24: 17.
Lingcon MH, Islam R, Khayer K, Islam MR. Cyclization of substituted indole-2-one-3-thiosemicarbazones to noble heterocyclic systems. J Bangladesh Chem Soc. 2001; 14: 127.
Islam R. Indian. J Chem. 2001; 40B: 240.
Kubota S, Yasufumi U, Fujikane K, Toyooka K, Shibuya M. Synthesis of 4-acyl-2-(acylamino)-D2-1,3,4-thiadiazolines and 4-acyl-2-amino-D2-1,3,4-thiadiazolines by acylation of thiosemicarbazones. J Org Chem. 1980; 45: 1473.
Islam MR, Khayer K, Islam R. Jahangirnagar Univ J Sci. 1997; 21: 1.
Microwell A. Cytotoxicity assay using Artemia salina (brine shrimp). Planta Med. 1997; 59: 250.

Apply citation style format of Bangladesh Journal of Pharmacology
Copyright (c) 2007 Md. Rabiul Islam, Mohammad Mohsin

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
